- BENZÉNOÏDES
- BENZÉNOÏDESL’adjectif substantivé «benzénoïde» désigne cette importante famille de composés de la chimie organique qui renferment, dans leur squelette carboné, l’enchaînement du noyau benzénique. Mono ou polycycliques, porteurs ou non de groupements fonctionnels hétéroatomiques ou de chaînes latérales aliphatiques ou alicycliques, les composés benzénoïdes présentent, en commun, un ensemble de propriétés spécifiques qu’ils doivent à l’existence, dans leur structure, d’un système électronique 神 particulièrement stable, et dont le benzène offre l’exemple le plus simple. L’ensemble de ces propriétés est lié à la notion d’aromaticité .La réactivité chimique particulière des composés benzénoïdes est caractérisée par la tendance de leur système électronique 神 à se reformer lorsque, dans une première phase de la réaction, il a été perturbé par l’attaque d’un réactif. Une conséquence de cette stabilité remarquable du système électronique 神 est que les composés benzénoïdes entrent normalement dans des réactions de substitution, alors que les composés, insaturés comme eux, mais non benzénoïdes, subissent de préférence des réactions d’addition. L’addition elle-même revêt, en série benzénoïde, un aspect particulier.Qu’ils soient extraits des produits de la pyrogénation des charbons ou qu’ils résultent de traitements thermiques et catalytiques des pétroles, les hydrocarbures benzénoïdes jouent un rôle fondamental dans l’économie moderne: par la rigidité et la stabilité de leur structure, ils interviennent dans l’élaboration de fibres textiles synthétiques, de polymères et d’élastomères présentant de remarquables qualités mécaniques et thermiques; par la facile excitation de leur système électronique 神, ils constituent les matières premières de choix de l’industrie des colorants synthétiques; par la facilité avec laquelle ils peuvent être substitués, ils sont utilisés dans la fabrication des détergents, des pesticides et de nombreux produits pharmaceutiques. Certains d’entre eux sont enfin d’excellents solvants.1. GénéralitésLes composés benzénoïdes renferment dans leur molécule un ou plusieurs cycles benzéniques. Ils appartiennent à la classe des squelettes dits alternants et présentent l’ensemble des caractères liés à l’aromaticité.On distingue les hydrocarbures benzénoïdes dérivés du benzène par un remplacement d’un ou de plusieurs atomes d’hydrogène par autant de chaînes hydrocarbonées et les dérivés fonctionnels de ces hydrocarbures, dans la structure desquels interviennent un ou plusieurs hétéroatomes (halogènes, oxygène, soufre, azote...).Les squelettes fondamentaux des hydrocarbures benzénoïdes (tabl. 1) peuvent être divisés en deux catégories:– le benzène et ses homologues mono ou polycycliques à noyaux séparés (dont les cycles ne possèdent aucun atome en commun);– les hydrocarbures polycycliques à noyaux condensés, dont les cycles possèdent en commun deux atomes de carbone adjacents.Nomenclature. IsomérieLa plupart des hydrocarbures benzénoïdes sont désignés par des noms courants également utilisés dans la nomenclature de leurs dérivés fonctionnels. Certains de ces derniers portent des noms courants, dont quelques-uns sont indiqués dans le tableau 2.Lorsqu’il intervient comme substituant par l’un de ses sommets, le noyau benzénique est désigné par le préfixe phényle, le noyau naphtalénique par le préfixe naphtyle ( 見 ou 廓). Lorsque deux des sommets sont engagés dans la substitution, ces préfixes deviennent phénylène ou naphtylène.L’isomérie de position des dérivés disubstitués du benzène se traduit, dans leur nomenclature, par les préfixes ortho- (-1,2), méta- (-1,3) ou para- (-1,4); dans le cas du naphtalène, les substitutions en positions -1, -4, -5 ou -8 correspondent à la lettre 見, en positions -2, -3, -6 ou -7 à la lettre 廓; il en est de même pour l’anthracène où les positions -9 et -10 sont désignées par le préfixe méso- . Les sommets -1,8 (ou -4,5) du cycle naphtalénique sont situés en position péri- .Structure électroniqueLa structure électronique des hydrocarbures benzénoïdes est caractérisée par une conjugaison cyclique de leur système électronique 神. Responsable de l’ensemble des propriétés spécifiques de ces hydrocarbures et de leurs dérivés fonctionnels, cette conjugaison cyclique se manifeste de diverses manières:Sur le plan énergétique , d’une part, par une stabilité remarquable du cycle aromatique, l’énergie de conjugaison est beaucoup plus importante pour le benzène (150 kJ . mole-1), le naphtalène (322 kJ . mole-1), l’anthracène (485 kJ . mole-1) et leurs homologues, que pour les systèmes conjugués ouverts (quelques kJ . mole-1), d’autre part, par une facile excitation du système électronique 神: les composés benzénoïdes sont de puissants chromophores.Sur le plan structural , par une rigidité du squelette carboné, qui est plan et dont les liaisons entre atomes de carbone ont des longueurs très voisines (0,139 nm pour le benzène, de 0,137 à 0,143 nm pour le naphtalène). Cette rigidité du squelette aromatique impose une orientation relative fixe des liaisons qu’il échange avec les groupes substituants.Sur le plan électrique , par l’assimilation du système électronique 神 à un petit circuit électrique, fermé, qu’il est possible de perturber localement par une substitution ou globalement par l’application d’un champ magnétique. Dans le premier cas, la perturbation se transmet, le long du circuit, aux autres sommets du cycle, dans le second, le courant induit, appelé courant de cycle, crée, au niveau des atomes d’hydrogène du noyau, un champ magnétique caractéristique des systèmes aromatiques. Une particularité des hydrocarbures benzénoïdes alternants est la répartition uniforme de la charge électronique 神 portée par leurs différents sommets, et la transmission alternée d’une perturbation subie par l’un d’entre eux.Sur le plan chimique , par la tendance que présente le système conjugué cyclique à se reformer au cours de réactions où, temporairement, il aurait été désorganisé; de cette stabilité remarquable du système 神 résulte la possibilité, pour les composés aromatiques, d’entrer dans des réactions de substitution; par la transmission, le long du système cyclique des électrons 神, des perturbations électroniques, apportées par un substituant placé sur un sommet, un substituant oriente les substitutions ultérieures; par la réactivité particulièrement élevée des chaînes latérales hydrocarbonées au niveau de l’atome de carbone directement lié au cycle: dans ce cas, en effet, l’intermédiaire de la réaction peut présenter une conjugaison supplémentaire (accompagnée de stabilisation) entre le centre réactif et le système 神 du cycle; par la modification des propriétés fondamentales des groupements fonctionnels directement fixés sur le cycle, en raison d’une conjugaison entre ce dernier et un doublet p ou 神 porté par l’atome de ce groupe lié au cycle.2. Propriétés physiquesLes domaines dans lesquels les composés aromatiques présentent des propriétés physiques spécifiques sont les suivants:Absorption ultravioletteEn raison de sa conjugaison cyclique, le système électronique 神 de ces composés est plus facile à déformer que celui des doubles liaisons isolées et même des systèmes ouverts (tabl. 3). L’absorption propre du système aromatique se situe dans le proche ultraviolet et l’extension de la conjugaison par introduction, dans le système, de substituants appropriés, comme le groupe nitro- (tabl. 2, formule 16 d) ou azoïque (tabl. 2, formule 23), provoque le déplacement (bathochrome) de l’absorption dans le spectre visible. On dit, pour cette raison, que les squelettes benzénoïdes sont des chromophores et que les substituants fonctionnels conjugués sont des auxochromes . La structure fine de la bande d’absorption, de plus grande longueur d’onde et de faible intensité, appelée bande benzénoïde (tabl. 3, ) est assez complexe et caractéristique des structures aromatiques (structure vibrationnelle).Absorption infrarougeLa nature particulière des liaisons des squelettes benzénoïdes entraîne pour ces derniers l’apparition dans leur spectre d’absorption infrarouge (et de diffusion Raman) de bandes caractéristiques qui permettent leur identification. On note en particulier deux bandes dans le domaine 1 500-1 600 cm-1 dues à des vibrations d’élongation 益 C-C et la bande située vers 3 030 cm-1 due aux vibrations d’élongation 益 C-H du cycle.Absorption en résonance magnétique nucléaireLes atomes d’hydrogène portés par les cycles aromatiques résonnent aux champs faibles en comparaison de ceux que portent les systèmes conjugués ouverts. La cause principale de cette différence réside dans l’existence, pour les systèmes benzénoïdes, d’un courant de cycle qui crée, au niveau des protons de ces systèmes, un petit champ qui s’ajoute au champ extérieur et augmente ainsi le champ local. Ce déblindage propre aux systèmes aromatiques est utilisé en analyse structurale pour identifier les «protons aromatiques». L’analyse des couplages de spin JH-H permet également de distinguer entre isomères de positions: JH-H ortho- = 5 à 8 Hz; JH-H méta- = 1 à 3 Hz; JH-H para- = 0 à 1 Hz.Le couplage de spin nucléaire entre un proton et l’isotope 13C du carbone aromatique auquel il est lié (la concentration dans la nature de 13C qui est de 1 p. 100 permet parfois de l’observer sans enrichissement) présente une valeur caractéristique de ces systèmes (J 13CH = 159 Hz pour le benzène).Spectrométrie de masseL’importante stabilisation due à la conjugaison cyclique du système électronique 神 des molécules benzénoïdes entraîne une difficulté relative des processus de fragmentation de leurs cycles. La conjugaison propre aux cations du type du benzylium, C6H5 漣 CH2+, confère à ces fragments une stabilité particulière qui les fait régulièrement apparaître dans le spectre de masse des composés de structure C6H5 漣 CH2 漣 R, à la position m /e = 91, sous la forme particulièrement stable d’ions tropylium. D’une manière générale, le spectre d’un composé benzénoïde, dont la formule est R 漣 C6H4 漣 CH2 漣 R, contient le fragment cationique R 漣 C7H6+.Les structures polycycliques, à cycles indépendants ou condensés, présentent également une faible fragmentation au niveau de leurs cycles.3. Propriétés chimiquesLe système électronique des composés benzénoïdes est caractérisé par la présence, autour d’un squelette formé de liaisons 靖, d’un système d’électrons 神, moins solidement liés énergétiquement: c’est ce système 神 qui interviendra dans les réactions chimiques. Les réactifs susceptibles d’attaquer ces composés sont donc essentiellement électrophiles (avides d’électrons). Ce sont soit des cations (chargés positivement), soit des molécules polaires présentant un centre électroniquement désaturé, soit des molécules polarisables (susceptibles de présenter, à l’approche du substrat aromatique, un centre électroniquement désaturé). Les radicaux libres sont également susceptibles de se combiner au système 神 de ces substrats.L’orientation de la substitution électrophile sur le substrat aromatique sera déterminée à la fois par la charge des différents sommets et par la facilité avec laquelle un doublet 神 peut être mis à la disposition du réactif au niveau de chacun d’eux. Le premier critère n’intervient pratiquement que dans le cas des dérivés fonctionnels des hydrocarbures benzénoïdes, dont la répartition électronique n’est pas uniforme (perturbations dues aux hétéroatomes), il n’a que peu d’importance pour les hydrocarbures euxmêmes, dont la charge est sensiblement unitaire au niveau de tous les sommets. Le second critère exprime la différence de polarisabilité du système électronique 神 au niveau des différents atomes de carbone du squelette: il est seul à intervenir dans le cas de la substitution radicalaire (radicaux libres électriquement neutres).Mécanisme général de la substitution électrophile sur les substrats aromatiquesL’étude cinétique de cette réaction a permis d’établir un schéma général pour décrire son mécanisme (tabl. 4). Supposons que le réactif électrophile soit un cation E+ qui attaque une molécule de toluène. Ce cation s’approche du substrat selon une direction perpendiculaire au plan du cycle; c’est en effet dans cette direction que la densité du système 神 est la plus grande et que sa polarisabilité est la plus importante. Il se forme, dans un premier temps, un complexe peu solide entre le substrat et le réactif E+, appelé complexe 神 (tabl. 4, b). Ce complexe 神 se réorganise en localisant le réactif E au niveau de l’atome de carbone du substrat vers lequel le transfert d’un doublet 神 est énergétiquement le plus favorable (sommet le plus polarisé ou le plus polarisable), et forme un nouveau complexe, appelé complexe 靖 (tabl. 4, c), dans lequel l’atome de carbone, centre de réaction, est lié par une liaison 靖 au réactif E. Le système 神 de ce complexe, ne comportant plus que deux doublets, répartis sur les cinq atomes de carbone restants, a perdu son caractère de conjugaison cyclique et l’importante stabilisation qui lui est attachée: la durée de vie du complexe 靖 peut être extrêmement courte (de l’ordre d’une période de vibration de liaison), c’est alors un état de transition, ou bien elle peut être suffisante pour lui donner une existence transitoire, et c’est alors un complexe intermédiaire.Dans les deux cas, le complexe 靖 se transforme spontanément en éliminant un proton H+ pour restaurer le système conjugué cyclique d’électrons 神, en formant le produit final de la réaction (tabl. 4, e). L’élimination du proton peut se faire par l’intermédiaire d’un nouveau complexe 神 (tabl. 4, d). Dans certains cas (réactions réversibles), le complexe 靖 peut éliminer le cation E+ et on retrouve alors le substrat de départ. Généralement, le proton est éliminé par l’anion qui accompagne le réactif cationique E+. Un mécanisme semblable faisant intervenir un complexe 靖 a été proposé pour décrire la réaction de substitution des composés benzénoïdes par les radicaux libres R. (tabl. 5). Un radical libre R . est ici nécessaire pour décomposer le complexe 靖 en donnant le produit final de substitution.Règles d’orientationCertaines réactions de substitution électrophile sont irréversibles (pour un domaine particulier de température): c’est le cas de la nitration et de l’halogénation. La substitution se produit alors sur le sommet le plus réactif (celui qui correspond à la plus petite énergie de localisation, à son niveau, d’un doublet 神 du cycle) et on dit que la réaction présente une orientation cinétique: le produit principal de la réaction est celui qui se forme le plus rapidement.D’autres réactions sont réversibles, comme la sulfonation (à température suffisamment élevée), l’alkylation et l’acylation de Friedel et Crafts; dans ce cas le produit final le plus abondant est celui qui est le plus stable, car, par suite de la réversibilité de la réaction, le système évolue vers l’état énergétiquement le plus favorable et on dit que la réaction présente une orientation thermodynamique. L’orientation de la substitution électrophile (irréversible) sur les différents sommets d’un substrat benzénique monosubstitué C6H5 漣 X dépend, comme on l’a vu, de l’influence exercée par le substituant X déjà présent sur la polarisabilité électrophile des cinq autres sommets. Le résultat de la réaction est en général un mélange des trois isomères possibles, obtenus dans des proportions variables selon la nature du groupement X.Le tableau 6 groupe quelques résultats relatifs à la nitration. On note, d’une part, les pourcentages d’isomères, d’autre part, la réactivité globale du substrat relativement à celle du benzène et enfin les facteurs partiels de vitesse f i pour chacune des positions du substrat. Chaque facteur partiel de vitesse f i représente le rapport de la vitesse de réaction du sommet i à celle d’un sommet du benzène. La comparaison entre eux des facteurs partiels de vitesse d’un même substrat permet de préciser l’orientation de la réaction; la valeur, relative à l’unité, de chacun d’eux permet en outre de comparer la réactivité du sommet correspondant à celle d’un sommet benzénique (activation pour f i 礪 1, désactivation pour f i 麗 1). L’expérience montre (et les calculs théoriques prévoient) que les substituants X peuvent être classés en trois groupes:– ceux qui activent le cycle (par rapport au benzène) et orientent la réaction essentiellement en ortho - et en para - de X; ce sont principalement: 漣-, 漣OR, 漣NR2 (R = H, alkyle), R (alkyle);– ceux qui désactivent le cycle, mais orientent la réaction essentiellement en ortho - et en para - de X; ce sont principalement les halogènes et les groupes halométhyle: face=F0019 漣Cl, 漣Br, 漣I, 漣CH2Cl...;– ceux qui désactivent le cycle et orientent la réaction essentiellement en méta -de X; ce sont principalement les groupes fortement électron-attracteurs et ceux qui possèdent des doublets 神 avec lesquels le cycle peut se conjuguer: face=F0019 漣 C2R, 漣 COR, 漣 2, 漣 C3, 漣 NR3+.Ces règles d’orientation, qui avaient été établies empiriquement (Hollemann), ont trouvé une justification théorique dans le calcul des énergies de localisation, pour la substitution électrophile, fondé sur le mécanisme développé plus haut (complexe 靖 ou complexe de Wheland).NitrationLe remplacement d’un atome d’hydrogène d’un composé aromatique par un groupement nitré (face=F0019 漣 2) est généralement réalisé par l’action d’un mélange, en proportions convenables, d’acides nitrique et sulfurique concentrés. Le bilan de la réaction peut s’écrire:
 où C6H5 漣 H symbolise le substrat aromatique.Le rôle de l’acide sulfurique est essentiellement de transformer l’acide nitrique en un réactif fortement électrophile, l’ion nitronium 2+:
où C6H5 漣 H symbolise le substrat aromatique.Le rôle de l’acide sulfurique est essentiellement de transformer l’acide nitrique en un réactif fortement électrophile, l’ion nitronium 2+: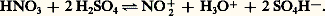 D’autres réactifs peuvent jouer un rôle analogue: l’acide nitrique lui-même (acide nitrique concentré), l’anhydride ou l’acide acétique, le nitrométhane. Les nitrates de benzoyle ou d’acétyle sont également employés comme agents de nitration.Étant irréversible, la nitration présente une orientation cinétique: le toluène, par exemple, donne successivement un mélange de 2-nitrotoluène et de 4-nitrotoluène puis le 2,4-dinitrotoluène et le 2,4,6-trinitrotoluène. L’introduction du premier groupement nitrodésactive le substrat et le mononitrotoluène est plus difficile à nitrer que le toluène, il en est de même du dinitrotoluène, plus difficile à nitrer que le mono-. Pour cette raison on utilise industriellement l’acide sulfonitrique résiduaire de la troisième nitration (dilué par l’eau formée dans la réaction) pour réaliser la seconde et l’acide résiduaire de la seconde pour réaliser la première. Le naphtalène est facilement nitré en 1-nitronaphtalène. Les dérivés nitrés aromatiques sont d’importantes matières intermédiaires de la fabrication des matières colorantes, certains d’entre eux sont des explosifs traditionnels (tolite ou trinitro-2,4,6 toluène: T.N.T.).NitrosationL’introduction d’un groupement nitroso 漣 N=O par substitution électrophile n’est pratiquement réalisable que sur des substrats particulièrement activés, comme les phénols, les éthers phénoliques, les amines tertiaires. L’agent de nitrosation usuel est l’acide nitreux ou le peroxyde d’azote. Les dérivés nitrosés sont des intermédiaires de synthèse intéressants.Couplage azoïqueCette réaction est très importante dans la préparation de matières colorantes synthétiques: le groupement azoïque 漣 N 略N 漣 est en effet un chromophore très puissant, et les composés azoïques aromatiques sont colorés. L’introduction du groupe fonctionnel azoïque résulte de l’attaque de substrats benzénoïdes très activés (amines aromatiques et phénols) par les sels d’aryl diazonium C6H5 漣 + 令 N,X- (X-: anion chlorure, sulfate...), qui sont des réactifs faiblement électrophiles. La réaction appelée couplage azoïque est fortement influencée par le pH de la solution dans laquelle elle a lieu: dans le cas où le substrat est un phénol, le pH le plus favorable est compris entre 8 et 11, dans celui où le substrat est une amine aromatique, le pH peut être plus faible. Les substituants, électro-attracteurs, comme 漣 2, 漣 Cl, augmentent la réactivité du réactif diazonium, les substituants électro-donneurs, comme 漣 OCH3, la diminuent au contraire. Les matières colorantes azoïques renferment en outre, dans leur molécule, des groupements auxiliaires qui leur confèrent une affinité pour les fibres textiles naturelles ou synthétiques.SulfonationC’est la réaction qui permet d’introduire un groupement acide sulfonique 漣 S3H sur un substrat aromatique. L’étude cinétique de la sulfonation a montré que le principal réactif de sulfonation est l’acide conjugué H3S4+ dans les solutions aqueuses concentrées d’acide sulfurique, et S3 dans les oléums, formés selon l’équilibre:
D’autres réactifs peuvent jouer un rôle analogue: l’acide nitrique lui-même (acide nitrique concentré), l’anhydride ou l’acide acétique, le nitrométhane. Les nitrates de benzoyle ou d’acétyle sont également employés comme agents de nitration.Étant irréversible, la nitration présente une orientation cinétique: le toluène, par exemple, donne successivement un mélange de 2-nitrotoluène et de 4-nitrotoluène puis le 2,4-dinitrotoluène et le 2,4,6-trinitrotoluène. L’introduction du premier groupement nitrodésactive le substrat et le mononitrotoluène est plus difficile à nitrer que le toluène, il en est de même du dinitrotoluène, plus difficile à nitrer que le mono-. Pour cette raison on utilise industriellement l’acide sulfonitrique résiduaire de la troisième nitration (dilué par l’eau formée dans la réaction) pour réaliser la seconde et l’acide résiduaire de la seconde pour réaliser la première. Le naphtalène est facilement nitré en 1-nitronaphtalène. Les dérivés nitrés aromatiques sont d’importantes matières intermédiaires de la fabrication des matières colorantes, certains d’entre eux sont des explosifs traditionnels (tolite ou trinitro-2,4,6 toluène: T.N.T.).NitrosationL’introduction d’un groupement nitroso 漣 N=O par substitution électrophile n’est pratiquement réalisable que sur des substrats particulièrement activés, comme les phénols, les éthers phénoliques, les amines tertiaires. L’agent de nitrosation usuel est l’acide nitreux ou le peroxyde d’azote. Les dérivés nitrosés sont des intermédiaires de synthèse intéressants.Couplage azoïqueCette réaction est très importante dans la préparation de matières colorantes synthétiques: le groupement azoïque 漣 N 略N 漣 est en effet un chromophore très puissant, et les composés azoïques aromatiques sont colorés. L’introduction du groupe fonctionnel azoïque résulte de l’attaque de substrats benzénoïdes très activés (amines aromatiques et phénols) par les sels d’aryl diazonium C6H5 漣 + 令 N,X- (X-: anion chlorure, sulfate...), qui sont des réactifs faiblement électrophiles. La réaction appelée couplage azoïque est fortement influencée par le pH de la solution dans laquelle elle a lieu: dans le cas où le substrat est un phénol, le pH le plus favorable est compris entre 8 et 11, dans celui où le substrat est une amine aromatique, le pH peut être plus faible. Les substituants, électro-attracteurs, comme 漣 2, 漣 Cl, augmentent la réactivité du réactif diazonium, les substituants électro-donneurs, comme 漣 OCH3, la diminuent au contraire. Les matières colorantes azoïques renferment en outre, dans leur molécule, des groupements auxiliaires qui leur confèrent une affinité pour les fibres textiles naturelles ou synthétiques.SulfonationC’est la réaction qui permet d’introduire un groupement acide sulfonique 漣 S3H sur un substrat aromatique. L’étude cinétique de la sulfonation a montré que le principal réactif de sulfonation est l’acide conjugué H3S4+ dans les solutions aqueuses concentrées d’acide sulfurique, et S3 dans les oléums, formés selon l’équilibre: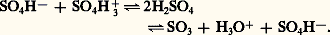 La réaction de sulfonation est réversible, mais cette réversibilité ne se manifeste en pratique que pour des températures suffisamment élevées et éventuellement en présence d’acide sulfurique moyennement concentré. Le naphtalène, par exemple, est sulfoné à température suffisamment basse (de 0 à 100 0C) en position 見 (sommet le plus réactif) selon une orientation cinétique, tandis qu’à température plus élevée (de 140 à 160 0C) l’orientation est thermodynamique, et l’on obtient l’isomère 廓 (le plus stable). Il est d’ailleurs possible de transposer l’acide naphtalène 見-sulfonique en son isomère 廓 par chauffage à 180 0C dans de l’acide sulfurique (désulfonation suivie de sulfonation thermodynamiquement orientée). Cette réversibilité de la réaction est utilisée pour bloquer temporairement une position réactive d’un substrat aromatique, afin de diriger la substitution sur un autre sommet réactif ; il suffit, en fin d’opération, de désulfoner par traitement à l’acide sulfurique dilué et chaud pour remplacer, par un atome d’hydrogène, le groupement sulfonique.Les acides sulfoniques aromatiques sont des acides relativement forts, utilisés comme tels en chimie organique. Ils confèrent au substrat qui les porte une certaine solubilité dans l’eau (ionisation de la fonction 漣 S3H): à ce titre, ils interviennent comme groupements auxiliaires dans de nombreux intermédiaires d’intérêt industriel. Par fusion alcaline de leurs sels alcalins, ils donnent les phénates correspondants (fabrication des naphtols, de l’alizarine; tabl. 2, composé 22). La sulfonation intervient dans la fabrication des détergents anioniques du groupe des alkylarylsulfonates (comme le produit de sulfonation du dodécylbenzène; tabl. 1, composé 7): un avantage de ces détergents sulfoniques sur les savons (carboxyliques) est que leurs sels de calcium et de magnésium sont plus solubles dans l’eau.HalogénationLe mode d’introduction d’un atome d’halogène sur un substrat benzénoïde dépend de la nature de l’halogène et de celle du substrat. Le fluor est trop réactif pour servir à la préparation des dérivés fluorés, ces derniers sont obtenus par voie indirecte (réaction de Schiemann). Les dérivés chlorés peuvent être obtenus par action du chlore, généralement activé par un catalyseur acide (acide de Lewis comme FeCl3, ZnCl2, AlCl3). La bromation par le brome est normalement plus lente que la chloration et l’emploi de catalyseurs acides est général. Pour ces deux réactions, on doit éviter l’influence de la lumière qui provoque la coupure homolytique des halogènes et initie des halogénations radicalaires (substitution benzylique ou addition sur le cycle). L’iode étant plus faiblement électrophile que les autres halogènes, l’emploi d’un agent oxydant est nécessaire; le rôle de cet agent est de transformer l’iode moléculaire en un cation plus réactif.Les cinétiques d’halogénation sont assez complexes, elles indiquent cependant que la réaction (irréversible) procède par l’intermédiaire d’un complexe 靖. Le rôle du catalyseur acide est de polariser la molécule d’halogène en augmentant son pouvoir électrophile, selon la réaction:
La réaction de sulfonation est réversible, mais cette réversibilité ne se manifeste en pratique que pour des températures suffisamment élevées et éventuellement en présence d’acide sulfurique moyennement concentré. Le naphtalène, par exemple, est sulfoné à température suffisamment basse (de 0 à 100 0C) en position 見 (sommet le plus réactif) selon une orientation cinétique, tandis qu’à température plus élevée (de 140 à 160 0C) l’orientation est thermodynamique, et l’on obtient l’isomère 廓 (le plus stable). Il est d’ailleurs possible de transposer l’acide naphtalène 見-sulfonique en son isomère 廓 par chauffage à 180 0C dans de l’acide sulfurique (désulfonation suivie de sulfonation thermodynamiquement orientée). Cette réversibilité de la réaction est utilisée pour bloquer temporairement une position réactive d’un substrat aromatique, afin de diriger la substitution sur un autre sommet réactif ; il suffit, en fin d’opération, de désulfoner par traitement à l’acide sulfurique dilué et chaud pour remplacer, par un atome d’hydrogène, le groupement sulfonique.Les acides sulfoniques aromatiques sont des acides relativement forts, utilisés comme tels en chimie organique. Ils confèrent au substrat qui les porte une certaine solubilité dans l’eau (ionisation de la fonction 漣 S3H): à ce titre, ils interviennent comme groupements auxiliaires dans de nombreux intermédiaires d’intérêt industriel. Par fusion alcaline de leurs sels alcalins, ils donnent les phénates correspondants (fabrication des naphtols, de l’alizarine; tabl. 2, composé 22). La sulfonation intervient dans la fabrication des détergents anioniques du groupe des alkylarylsulfonates (comme le produit de sulfonation du dodécylbenzène; tabl. 1, composé 7): un avantage de ces détergents sulfoniques sur les savons (carboxyliques) est que leurs sels de calcium et de magnésium sont plus solubles dans l’eau.HalogénationLe mode d’introduction d’un atome d’halogène sur un substrat benzénoïde dépend de la nature de l’halogène et de celle du substrat. Le fluor est trop réactif pour servir à la préparation des dérivés fluorés, ces derniers sont obtenus par voie indirecte (réaction de Schiemann). Les dérivés chlorés peuvent être obtenus par action du chlore, généralement activé par un catalyseur acide (acide de Lewis comme FeCl3, ZnCl2, AlCl3). La bromation par le brome est normalement plus lente que la chloration et l’emploi de catalyseurs acides est général. Pour ces deux réactions, on doit éviter l’influence de la lumière qui provoque la coupure homolytique des halogènes et initie des halogénations radicalaires (substitution benzylique ou addition sur le cycle). L’iode étant plus faiblement électrophile que les autres halogènes, l’emploi d’un agent oxydant est nécessaire; le rôle de cet agent est de transformer l’iode moléculaire en un cation plus réactif.Les cinétiques d’halogénation sont assez complexes, elles indiquent cependant que la réaction (irréversible) procède par l’intermédiaire d’un complexe 靖. Le rôle du catalyseur acide est de polariser la molécule d’halogène en augmentant son pouvoir électrophile, selon la réaction: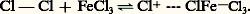 L’effet désactivateur du premier atome d’halogène, introduit sur un cycle aromatique, rend plus difficile la seconde halogénation.Les dérivés halogénés aromatiques sont peu réactifs, leur réactivité est toutefois augmentée par introduction, en ortho- ou para- de l’atome d’halogène, d’un groupement nitré. Ils sont utilisés comme intermédiaire de réactions et certains d’entre eux présentent d’intéressantes propriétés pesticides (p,p dichlorodiphényl-2,2 trichloroéthane, ou D.D.T.).Alkylation de Friedel et CraftsIl est possible d’obtenir des dérivés d’alkylation des substrats aromatiques par action, sur ces derniers, de réactifs électrophiles carbonés. Ces réactifs sont obtenus par action, sur des composés aliphatiques ou alicycliques porteurs d’une double liaison ou de groupes fonctionnels électro-attracteurs, d’acides protoniques forts ou d’acides de Lewis. Le rôle de l’acide est de transformer en réactif fortement électrophile l’agent d’alkylation:
L’effet désactivateur du premier atome d’halogène, introduit sur un cycle aromatique, rend plus difficile la seconde halogénation.Les dérivés halogénés aromatiques sont peu réactifs, leur réactivité est toutefois augmentée par introduction, en ortho- ou para- de l’atome d’halogène, d’un groupement nitré. Ils sont utilisés comme intermédiaire de réactions et certains d’entre eux présentent d’intéressantes propriétés pesticides (p,p dichlorodiphényl-2,2 trichloroéthane, ou D.D.T.).Alkylation de Friedel et CraftsIl est possible d’obtenir des dérivés d’alkylation des substrats aromatiques par action, sur ces derniers, de réactifs électrophiles carbonés. Ces réactifs sont obtenus par action, sur des composés aliphatiques ou alicycliques porteurs d’une double liaison ou de groupes fonctionnels électro-attracteurs, d’acides protoniques forts ou d’acides de Lewis. Le rôle de l’acide est de transformer en réactif fortement électrophile l’agent d’alkylation: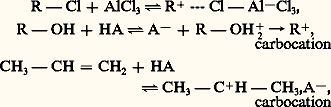 ou bien il polarise la liaison R 漣 Cl, ou bien il transforme le réactif en carbocation. Dans ce dernier cas, le carbocation formé peut se réarranger (transposition de Wagner-Meerwein). Il est difficile d’éviter la polyalkylation, par suite de l’activation apportée par le premier groupe alkyle fixé sur le substrat. La réaction d’alkylation de Friedel et Crafts peut être réversible en présence d’un excès de catalyseur, et des réarrangements (intraou intermoléculaires) se produisent.L’étude cinétique de cette réaction montre que l’intermédiaire de la réaction est un complexe du type 靖.Les applications pratiques de l’alkylation des hydrocarbures aromatiques sont nombreuses: préparation de l’éthylbenzène, dont la déshydrogénation catalytique donne le styrène (tabl. 1, formule 8), préparation du cumène à partir du benzène et du propylène (tabl. 1, formule 6), préparation du dodécylbenzène (tabl. 1, formule 7) à partir du tétramère du propylène.Acylation de Friedel et CraftsLe même type de réaction peut être réalisé à l’aide d’un réactif acylé: chlorure ou anhydride d’acide. Le catalyseur acide polarise fortement la liaison carbonyle et augmente le caractère électrophile de l’atome de carbone, la formation d’ions acylium est également possible. La cétone obtenue se trouve en général complexée avec le catalyseur si bien qu’un excès de ce dernier est nécessaire et qu’il faut hydrolyser le produit de la réaction pour libérer la cétone elle-même.L’acylation de Friedel et Crafts est largement utilisée en pratique. Un exemple intéressant d’application est la préparation de composés polycycliques.De nombreuses matières intermédiaires sont préparées par acylation de Friedel et Crafts de substrats benzénoïdes.Réactions d’additionQuelques réactions d’addition sont possibles en série benzénoïde, malgré l’incidence défavorable de la suppression de la conjugaison cyclique. La chloration du substrat symétrique du benzène se produit sur les six sommets lorsque cet hydrocarbure est soumis en phase liquide à l’action du chlore en présence de lumière bleue (chloration photochimique). Le produit de la réaction est un mélange d’isomères géométriques dont l’un (isomère 塚) possède de remarquables propriétés insecticides (hexachlorocyclohexane). L’hydrogénation du benzène, du phénol et de l’aniline se produit également sur les six sommets, par action de l’hydrogène en présence d’un catalyseur (Ni, Pt), en donnant respectivement le cyclohexane, le cyclohexanol et la cyclohexylamine. Dans le cas des dérivés polycycliques, la réduction procède par étapes. Sous l’action du sodium et de l’alcool, le naphtalène donne le dihydro-1,4 naphtalène, tandis que l’hydrogène, en présence de nickel, le transforme à 150 0C et sous 30 atm. en 1,2,3,4-tétrahydronaphtalène (tétraline) et à 200 0C et sous 200 atm. en décahydronaphtalène (décaline). L’hydrogénation catalytique de l’anthracène et du phénanthrène conduit à des dérivés dihydrogénés en positions -9,10.L’ozone s’additionne également sur les composés benzénoïdes donnant un tri-ozonide avec le benzène et un mono-ozonide avec le phénanthrène (positions -9,10).OxydationLes hydrocarbures aromatiques, sans chaîne latérale, sont oxydés par différents agents: l’oxydation du benzène est difficile, elle est réalisée par l’oxygène à 450 0C sur pentoxyde de vanadium et conduit à l’anhydride maléique; celle du naphtalène, plus facile, se produit sous l’action du permanganate en milieu alcalin, on obtient l’anhydride phtalique; celle de l’anthracène est encore plus facile, elle a lieu en présence du mélange acétochromique et donne l’anthraquinone. Il est intéressant de noter, à propos des dérivés monofonctionnels du naphtalène, qu’un groupe activateur en substitution (face=F0019 漣 NR2) rend fragile le cycle qui le porte tandis qu’un groupe désactivateur en substitution (face=F0019 漣 2) protège de l’oxydation le cycle qui le porte.Les chaînes alkylées subissent l’oxydation au niveau de l’atome de carbone fixé au cycle (position benzylique).4. Préparations. UtilisationsJusqu’au milieu du siècle environ, les hydrocarbures aromatiques industriels étaient obtenus comme sous-produits de la pyrogénation des charbons (1 000-1 300 0C) en vue de la fabrication du coke métallurgique ou du gaz d’éclairage (benzène, toluène, xylènes, naphtalène, méthyl-naphtalènes, anthracène...). Progressivement les pétroles remplacent le charbon comme source de ces carbures: les procédés d’aromatisation et de déhydrocyclisation des hydrocarbures alicycliques et aliphatiques permettent en effet de produire à bon compte le toluène, les xylènes et le benzène.La préparation au laboratoire d’hydrocarbures aromatiques fait appel soit à l’alkylation de Friedel et Crafts de substrats benzénoïdes, soit à l’acylation suivie de réduction et de déshydrogénation (cyclisation notamment); elle peut également se faire par l’intermédiaire de dérivés organo-métalliques aromatiques.Les hydrocarbures benzénoïdes et leurs dérivés fonctionnels ont une importance économique considérable; par leur stabilité, ils interviennent dans la fabrication de fluides de transfert thermique, de matériaux thermostables, d’élastomères résistants; par la rigidité de leur squelette, ils sont utilisés dans la confection de fibres textiles synthétiques, de mousses plastiques rigides, de résines thermodurcissables fines; par la facilité avec laquelle ils subissent des réactions de substitution, ils représentent des matières premières de choix pour l’introduction de groupements fonctionnels et interviennent dans la synthèse de médicaments, de pesticides, de solvants, de détergents; par leur caractère chromophore, ils sont à la base de l’industrie des matières colorantes de synthèse.
ou bien il polarise la liaison R 漣 Cl, ou bien il transforme le réactif en carbocation. Dans ce dernier cas, le carbocation formé peut se réarranger (transposition de Wagner-Meerwein). Il est difficile d’éviter la polyalkylation, par suite de l’activation apportée par le premier groupe alkyle fixé sur le substrat. La réaction d’alkylation de Friedel et Crafts peut être réversible en présence d’un excès de catalyseur, et des réarrangements (intraou intermoléculaires) se produisent.L’étude cinétique de cette réaction montre que l’intermédiaire de la réaction est un complexe du type 靖.Les applications pratiques de l’alkylation des hydrocarbures aromatiques sont nombreuses: préparation de l’éthylbenzène, dont la déshydrogénation catalytique donne le styrène (tabl. 1, formule 8), préparation du cumène à partir du benzène et du propylène (tabl. 1, formule 6), préparation du dodécylbenzène (tabl. 1, formule 7) à partir du tétramère du propylène.Acylation de Friedel et CraftsLe même type de réaction peut être réalisé à l’aide d’un réactif acylé: chlorure ou anhydride d’acide. Le catalyseur acide polarise fortement la liaison carbonyle et augmente le caractère électrophile de l’atome de carbone, la formation d’ions acylium est également possible. La cétone obtenue se trouve en général complexée avec le catalyseur si bien qu’un excès de ce dernier est nécessaire et qu’il faut hydrolyser le produit de la réaction pour libérer la cétone elle-même.L’acylation de Friedel et Crafts est largement utilisée en pratique. Un exemple intéressant d’application est la préparation de composés polycycliques.De nombreuses matières intermédiaires sont préparées par acylation de Friedel et Crafts de substrats benzénoïdes.Réactions d’additionQuelques réactions d’addition sont possibles en série benzénoïde, malgré l’incidence défavorable de la suppression de la conjugaison cyclique. La chloration du substrat symétrique du benzène se produit sur les six sommets lorsque cet hydrocarbure est soumis en phase liquide à l’action du chlore en présence de lumière bleue (chloration photochimique). Le produit de la réaction est un mélange d’isomères géométriques dont l’un (isomère 塚) possède de remarquables propriétés insecticides (hexachlorocyclohexane). L’hydrogénation du benzène, du phénol et de l’aniline se produit également sur les six sommets, par action de l’hydrogène en présence d’un catalyseur (Ni, Pt), en donnant respectivement le cyclohexane, le cyclohexanol et la cyclohexylamine. Dans le cas des dérivés polycycliques, la réduction procède par étapes. Sous l’action du sodium et de l’alcool, le naphtalène donne le dihydro-1,4 naphtalène, tandis que l’hydrogène, en présence de nickel, le transforme à 150 0C et sous 30 atm. en 1,2,3,4-tétrahydronaphtalène (tétraline) et à 200 0C et sous 200 atm. en décahydronaphtalène (décaline). L’hydrogénation catalytique de l’anthracène et du phénanthrène conduit à des dérivés dihydrogénés en positions -9,10.L’ozone s’additionne également sur les composés benzénoïdes donnant un tri-ozonide avec le benzène et un mono-ozonide avec le phénanthrène (positions -9,10).OxydationLes hydrocarbures aromatiques, sans chaîne latérale, sont oxydés par différents agents: l’oxydation du benzène est difficile, elle est réalisée par l’oxygène à 450 0C sur pentoxyde de vanadium et conduit à l’anhydride maléique; celle du naphtalène, plus facile, se produit sous l’action du permanganate en milieu alcalin, on obtient l’anhydride phtalique; celle de l’anthracène est encore plus facile, elle a lieu en présence du mélange acétochromique et donne l’anthraquinone. Il est intéressant de noter, à propos des dérivés monofonctionnels du naphtalène, qu’un groupe activateur en substitution (face=F0019 漣 NR2) rend fragile le cycle qui le porte tandis qu’un groupe désactivateur en substitution (face=F0019 漣 2) protège de l’oxydation le cycle qui le porte.Les chaînes alkylées subissent l’oxydation au niveau de l’atome de carbone fixé au cycle (position benzylique).4. Préparations. UtilisationsJusqu’au milieu du siècle environ, les hydrocarbures aromatiques industriels étaient obtenus comme sous-produits de la pyrogénation des charbons (1 000-1 300 0C) en vue de la fabrication du coke métallurgique ou du gaz d’éclairage (benzène, toluène, xylènes, naphtalène, méthyl-naphtalènes, anthracène...). Progressivement les pétroles remplacent le charbon comme source de ces carbures: les procédés d’aromatisation et de déhydrocyclisation des hydrocarbures alicycliques et aliphatiques permettent en effet de produire à bon compte le toluène, les xylènes et le benzène.La préparation au laboratoire d’hydrocarbures aromatiques fait appel soit à l’alkylation de Friedel et Crafts de substrats benzénoïdes, soit à l’acylation suivie de réduction et de déshydrogénation (cyclisation notamment); elle peut également se faire par l’intermédiaire de dérivés organo-métalliques aromatiques.Les hydrocarbures benzénoïdes et leurs dérivés fonctionnels ont une importance économique considérable; par leur stabilité, ils interviennent dans la fabrication de fluides de transfert thermique, de matériaux thermostables, d’élastomères résistants; par la rigidité de leur squelette, ils sont utilisés dans la confection de fibres textiles synthétiques, de mousses plastiques rigides, de résines thermodurcissables fines; par la facilité avec laquelle ils subissent des réactions de substitution, ils représentent des matières premières de choix pour l’introduction de groupements fonctionnels et interviennent dans la synthèse de médicaments, de pesticides, de solvants, de détergents; par leur caractère chromophore, ils sont à la base de l’industrie des matières colorantes de synthèse.
Encyclopédie Universelle. 2012.